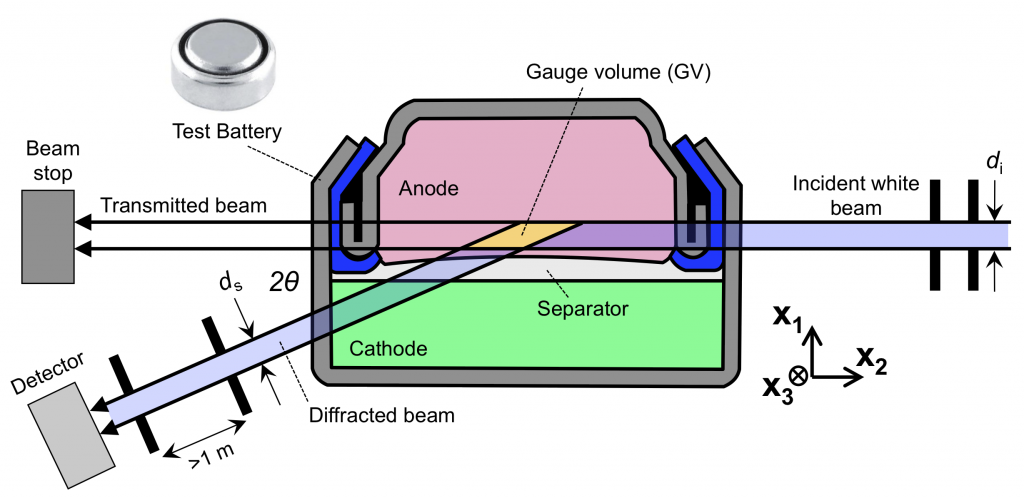
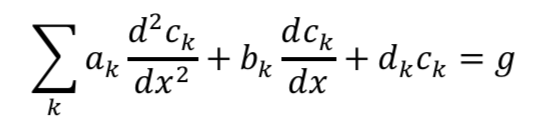We have a new paper out, which is a review paper on using energy dispersive X-ray diffraction (EDXRD) for battery characterization. EDXRD had previously been used to get crystallography data from inside diamond anvil cells, and also from the bulk interior of engineering materials like turbine blades. Using it to look inside sealed batteries was a good idea. It wasn’t my idea, but I’m one of the battery people they found to try it. I have a passion for current distributions, and EDXRD is a wonderful way to directly observe current distributions.
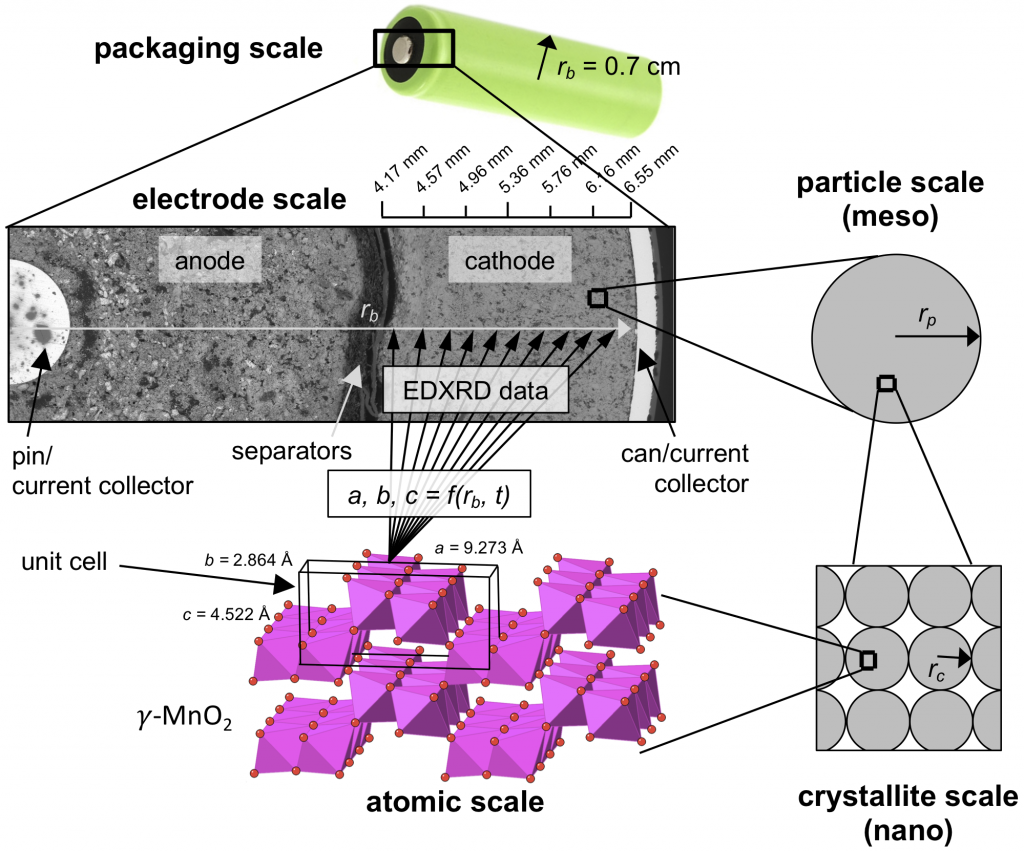
The paper was written with Amy Marschilok and the Takeuchi group at Stony Brook, and also with Mark Croft at Rutgers. We’re still using EDXRD to watch complex mechanisms inside batteries (without the fear that opening the battery will change anything), but since the review paper spans 2012-2020, it was a good idea to catalog past work in one place. The new HEX beamline under development at NSLS-II will be the premier EDXRD resource sometime soon. The figure below is an attempt to explain what exactly EDXRD gets you (atomic scale information, as a function of the electrode scale) and why it’s important (look how thick that electrode is). If you ever want to talk about EDXRD drop me a line.
As a model problem we’ll use the boundary value problem of Tafel kinetics in a concentration-independent porous electrode in Cartesian coordinates. The reason to choose this one is because Newman and Tobias provide the analytical solution in their 1962 paper “Theoretical Analysis of Current Distribution in Porous Electrodes.” You can also find this solution in Fuller and Harb’s Electrochemical Engineering, in the problems at the end of Chapter 5. The reason to choose a problem with an analytical solution is so you can check your work to know if it’s right! We lay out the problem below, which is the same as that in Newman and Tobias (1962) except one sign convention. This is four equations with four unknowns (i1, i2, Φ1, Φ2):
If you read the electrochemical engineering literature, solving problems such as this with Newman’s BAND method often comes up, and you might wonder what that is. Technically “BAND” is some Fortran code set up to solve 2-point boundary value problems in a way that is convenient for electrochemists. (My own favorite document written about BAND is “Modeling and reactor simulation” by Douglas Bennion in the AIChE symposium series 1983, Vol 79, Num 229, pp 25-36. It’s a resource meant for teachers and is somewhat hard to find so I’ve uploaded it here.) The formulation Newman uses to set up the problem casts each of the equations in this way:
Here c is a variable. The coefficients a, b, and d are those for the second derivative, first derivative, and the variable itself. And g is the constant term. When you linearize equation 2 above and then cast all of the four equations this way, you get something that looks like the tables below. I’ve collected some constants together as ß and P to keep it from looking too messy:
Once you have this, you can (a) build a block tridiagonal matrix, (b) choose initial guesses for the answers, and (c) iterate to a solution by repeatedly solving the problem until your initial guesses and final answers match. Explaining how this works is too long for a blog post, so follow these links for:
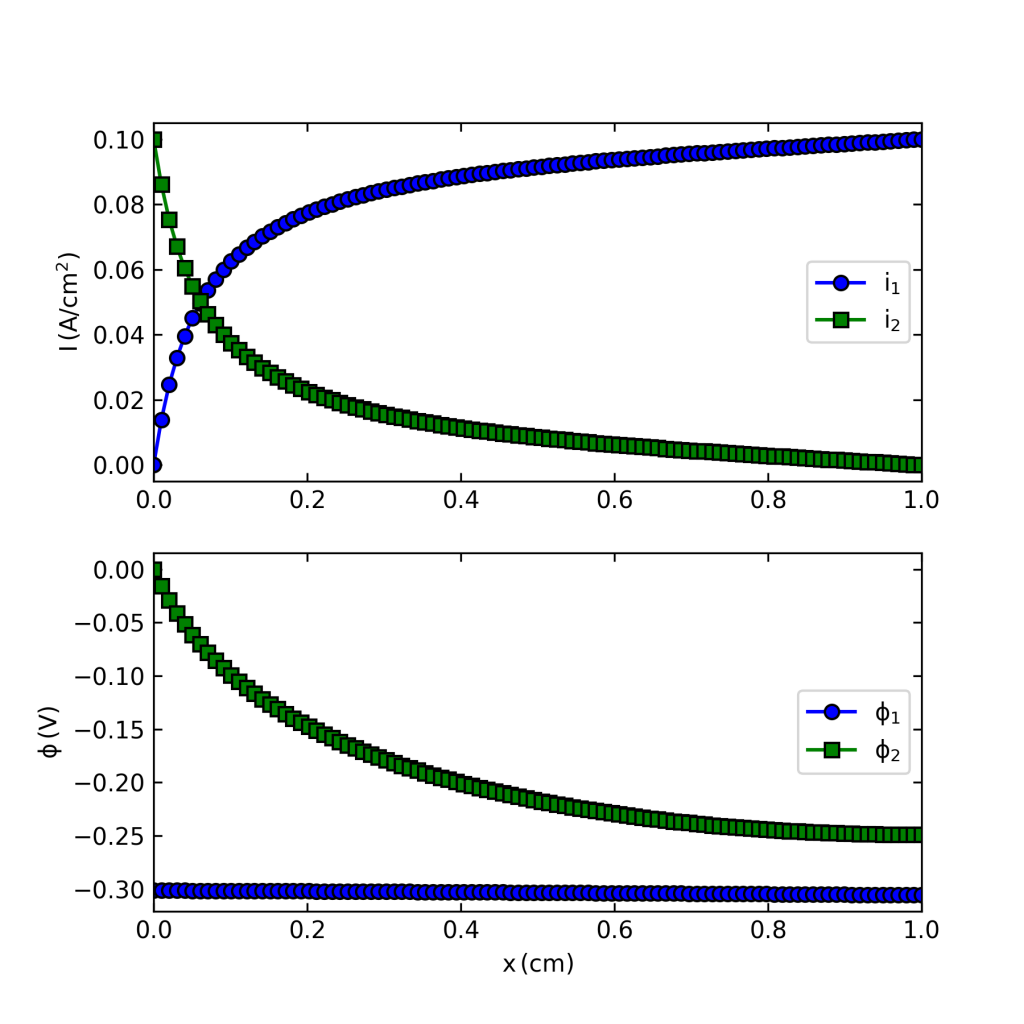
If you execute the code you should get a figure like that below, which solves the problem. (I still use Python 2.7 so you might have to do some mild editing and debugging.)
What I use this for
The reason I started writing a BAND-type method in Python is (1) I wanted to learn more about how BAND worked so I could teach it better, and (2) I wanted to model electrochemical systems without having to buy expensive software or use Fortran. It’s not exactly like Newman’s code, which also included a function to solve the matrix problem. Rather, this code lets you input the problem in Newman’s way and build the corresponding block tridiagonal matrix, then it uses SciPy to solve the matrix problem.
When I was working on the paper above in 2016, I wanted to compare experimental data to the Chen + Podlaha + Cheh alkaline battery model developed in the 1990s. Python seems like the best choice for something you can also develop as a teaching tool, so I used that to generate the results in Figure 3 of the paper. Since then I’ve used it to train some MS students getting insight into shallow-cycled MnO2 cathodes. When Zhicheng Lu’s MS thesis appeared on the internet, I received a few questions about the model by email. So this post is meant to help anyone doing similar things.
We have a new paper out, led by Dr. Damon Turney, which is required reading for anyone who wants to know the practical considerations for commercializing rechargeable zinc anodes, particularly energy density values. Inside there’s a modern literature survey of zinc alkaline anodes with levelized performance metrics, along with experimental assessment of leading formulations. Long-cycle materials characterization, performance metrics, and failure analysis are reported for over 25 unique anode formulations with up to 1500 cycles and ∼1.5 years of shelf life per test.
Some exciting news: we have a new paper out in Nature Communications featuring a deep cycling manganese dioxide (MnO2) cathode. This cathode is aqueous, operating for thousands of cycles in KOH electrolyte, at essentially the full 2-electron capacity of MnO2, which is 617 mAh/g-MnO2. MnO2 is abundant, inexpensive, and non-toxic, making it an ideal basis material for large-scale batteries at the scale of the power grid. Batteries on this scale will make widespread use of solar and wind power possible in the future. Because the electrolyte is water-based, the resulting battery is inherently safer than Li-ion, which relies on a highly flammable electrolyte. Fires caused by Li-ion batteries in small devices like cell phones are a generally accepted risk, but for massive stationary batteries located in power grid infrastructure the risk assessment recommends using a safer, water-based battery chemistry.
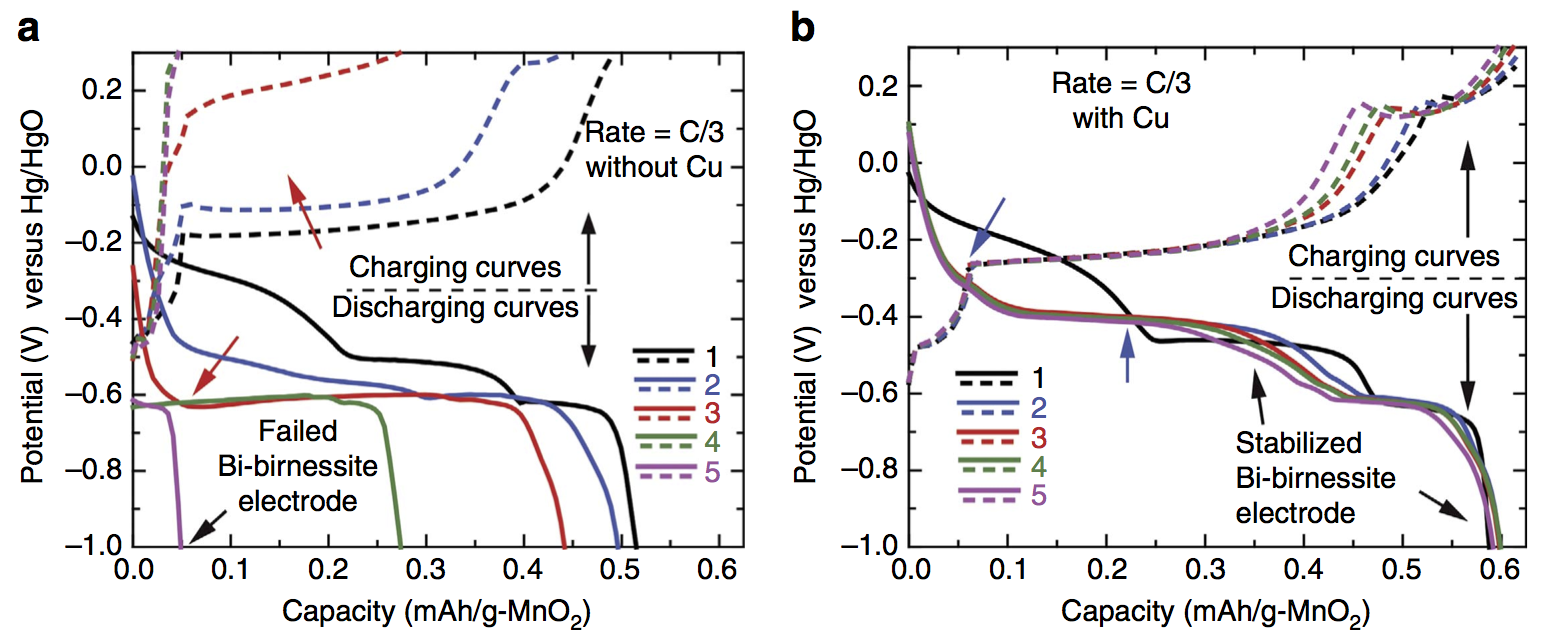
The paper title is Regenerable Cu-intercalated MnO2 layered cathode for highly cyclable energy dense batteries, and the innovation that makes everything possible is intercalating copper (Cu) into bismuth-modified δ-MnO2 (birnessite). Bismuth-birnessite was discovered by Ford Motor Company in the 1980s, but it was never known how to use it at high areal capacity. The first five cycles of a cathode with no copper (a) and with copper (b) are shown below. Without copper the cathode failed by the third cycle.
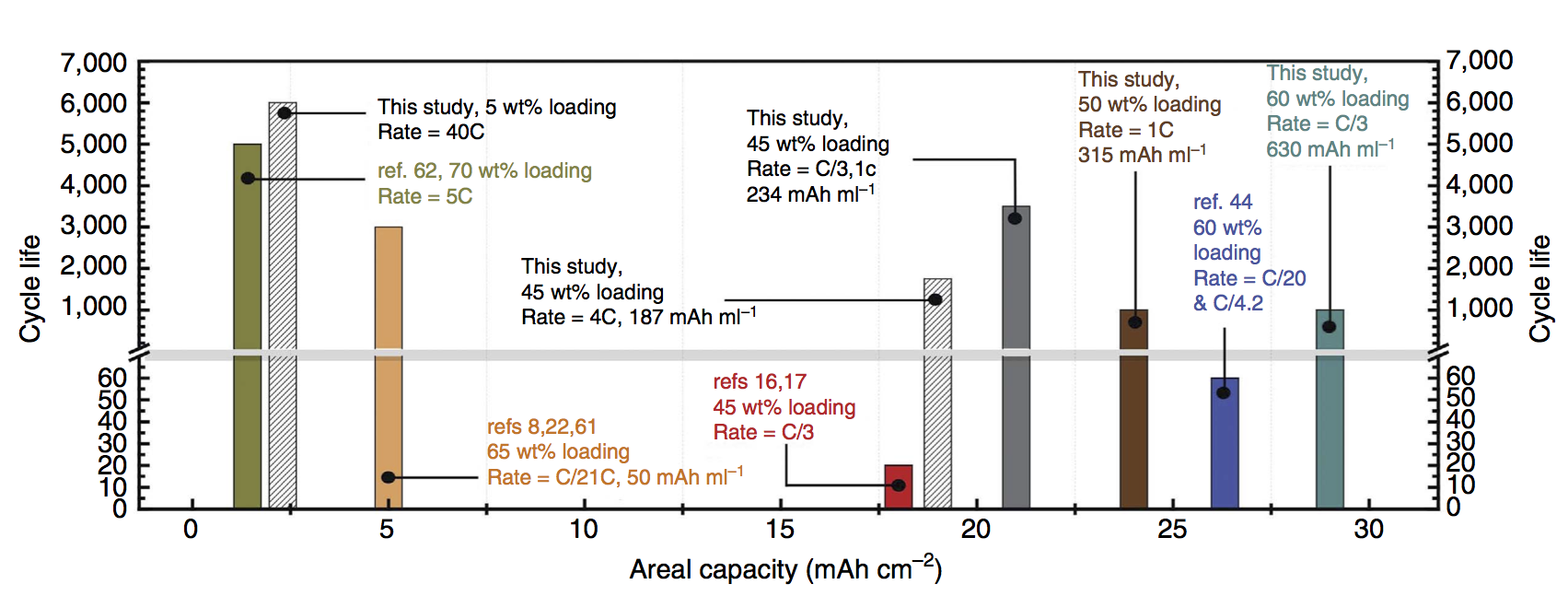
Gautam Yadav discovered this copper intercalation here at the CUNY Energy Institute during the final year of our ARPA-E project. The reason it is a big discovery is this: Bi-birnessite will cycle nicely under some conditions, for example at low mass loading or with a very thin electrode, i.e. situations with low areal capacity. In contrast, we achieved both high cycle life and very high areal capacity. Areal capacity is measured in mAh/cm2, and achieving high areal capacity is critical for packing a lot of electrodes together with high energy density. Any practical battery requires high areal capacity (as opposed to batteries meant as academic exercises or low-level proofs of concept).
The plot above puts areal capacity in context. One recent report (ref 62) achieved 5,000 cycles, but at a small areal capacity of 1 mAh/cm2. Another report (ref 44) successfully achieved 26 mAh/cm2, but with only 60 recharging cycles. By using Cu-intercalated MnO2 we achieved 6,000 cycles at 2.5 mAh/cm2 and 1,000 cycles at 28 mAh/cm2. Battery modeling suggests this opens up a design pathway to 200 Wh/L, equal to the energy density of some kinds of Li-ion battery, for an aqueous Zn-MnO2 battery.