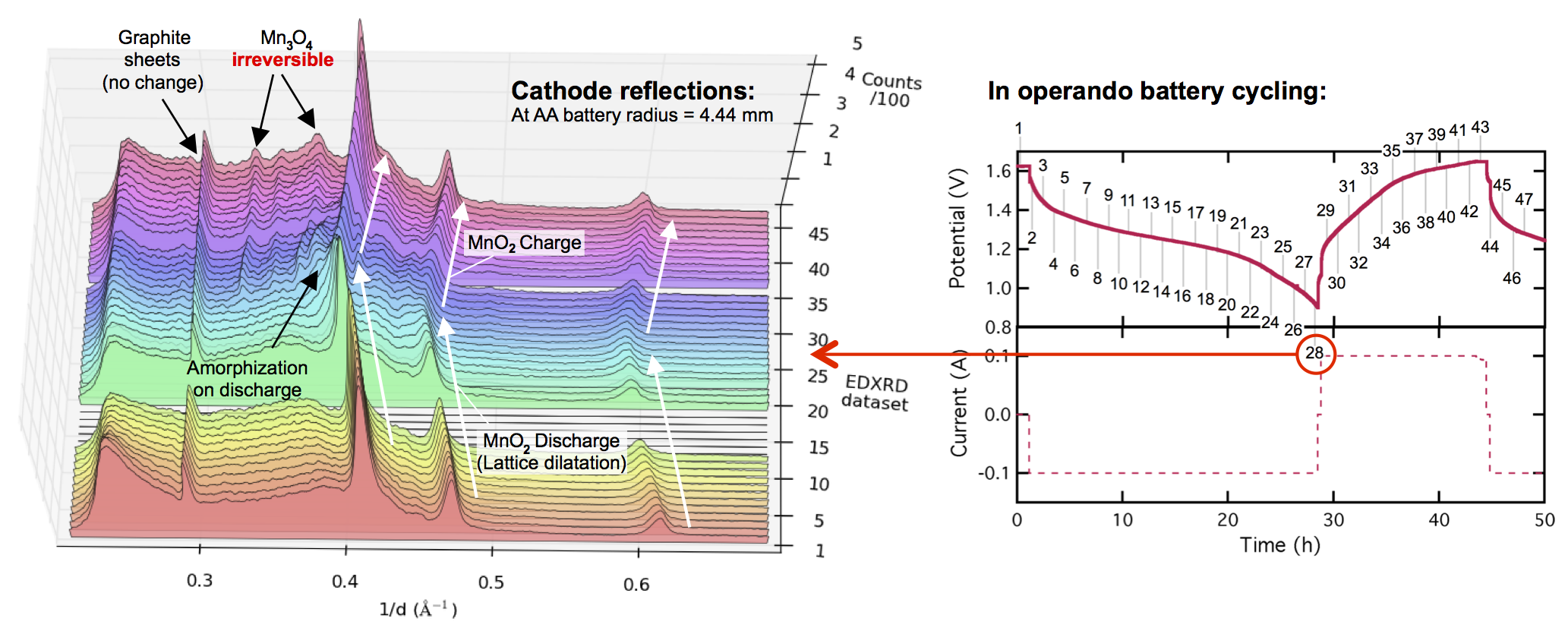
I’ve had quite a few people email asking what I think about the new paper Reversible aqueous zinc/manganese oxide energy storage from conversion reactions in Nature Energy, by authors at PNNL. Understandable, since I spend a lot of my time talking about Zn and MnO2 for electrical storage. Fair!

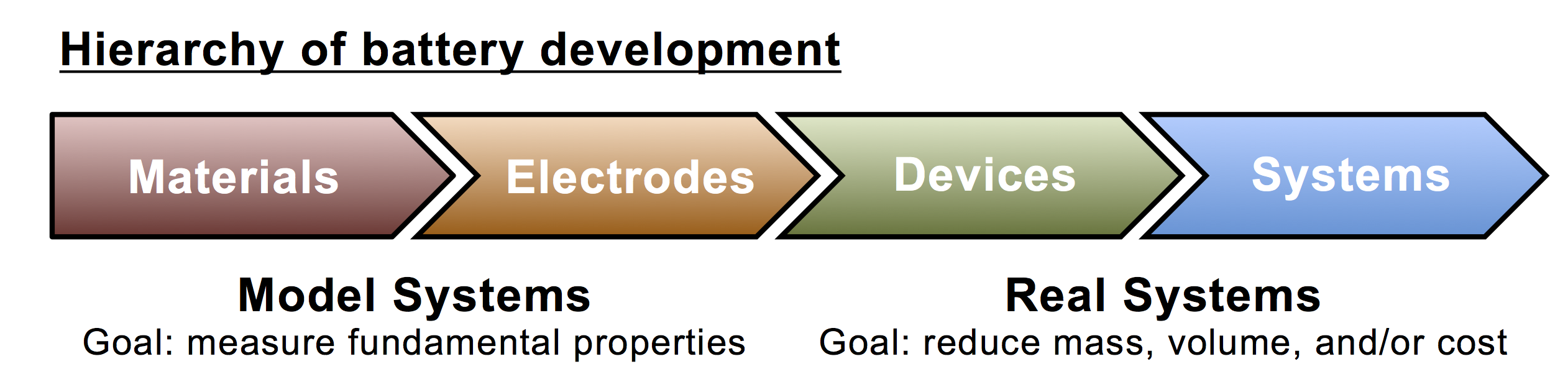
First: unfortunately the work is being marketed (by PNNL) with a terrible graphic of a Platonic ideal supergreen™ battery that sits in a sunlit field and emits rays of light that save the world, but that’s pretty standard for battery research these days. Once the PR department gets ahold of it, you’re waist-deep in pictures of suns, windmills, iPhones, and Teslas. Most people, even most scientists, don’t understand the many levels of hierarchy involved in battery design and engineering, so I try to overlook these kinds of silly Photoshop excursions.
Second: the innovation of the paper is that they are making a rechargeable Zn-MnO2 battery in a mildly acidic electrolyte, and getting good cycle life. The way they’re doing this is by using α-MnO2 as their cathode active material. MnO2 comes in several polymorphic forms, some of which you can see below. (I adapted this figure from a paper by Poinsignon.) They are built from MnO6 octahedra, but can distinguished by the tunnel structures in the crystal.
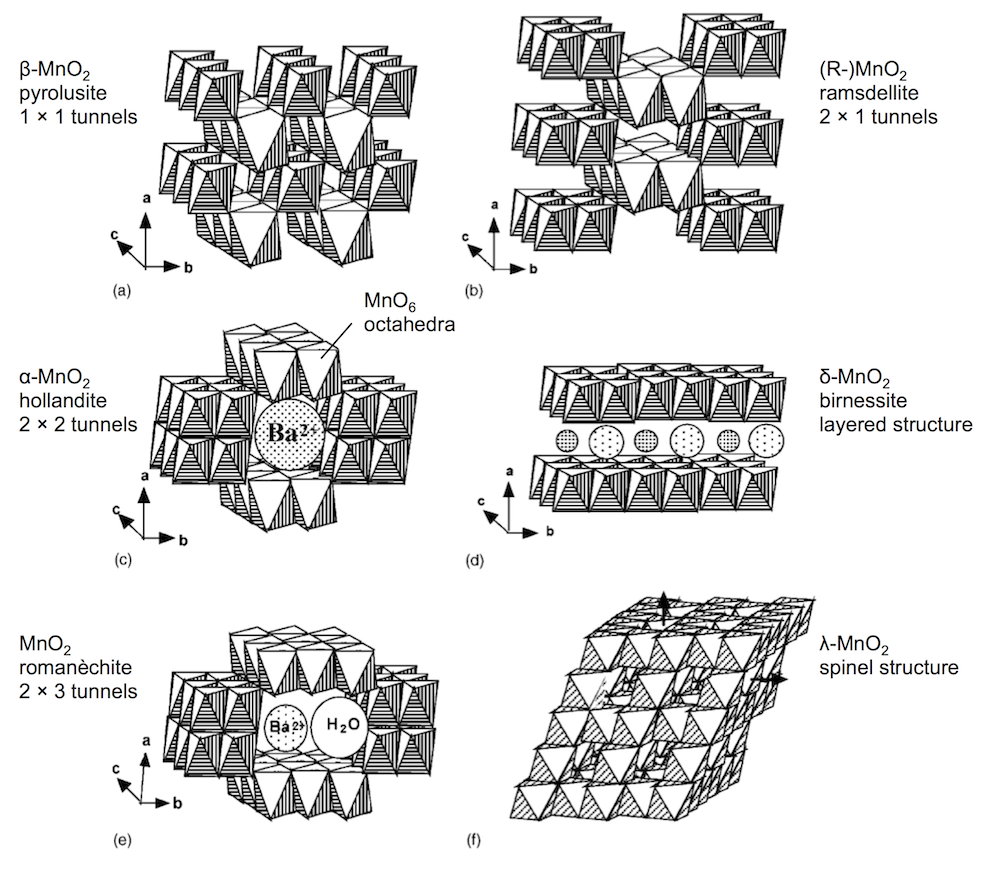
A lot of my recent work has focused on the polymorph γ-MnO2, which is an intergrowth of (a) and (b) above. The PNNL work makes an interesting discovery about α-MnO2: they see the α-MnO2 going through a conversion reaction to MnOOH, which is somewhat unexpected. As you can see in the figure above, α-MnO2 is usually thought of as a host structure, to intercalate guest ions (like Ba2+). They then see that the surface of the MnOOH is coated with a large flake-like material that originates with the sulfate electrolyte, ZnSO4[Zn(OH)2]3 · xH2O. In this respect, the reaction is a bit reminiscent of a lead-acid battery, which also involves a sulfate film.
The paper is very interesting in that it provides unexpected evidence of α-MnO2 acting in the manner of a conversion reaction. (And that’s why that term is important and shows up in the paper’s title.) Also the zinc hydroxyl sulfate flake-film is a tantalizing look at what could be a very complex cathode reaction. And I’m a sucker for complex electrochemical reactions, as I hope you know. The test bed for the research was a CR2032 form factor, which is the kind of battery that goes in my running watch. So, the picture the PR machine and the science press are painting (with that world-saving battery up above) is a bit overblown, but the electrochemistry research underpinning this paper is extremely interesting, and I hope to see more.